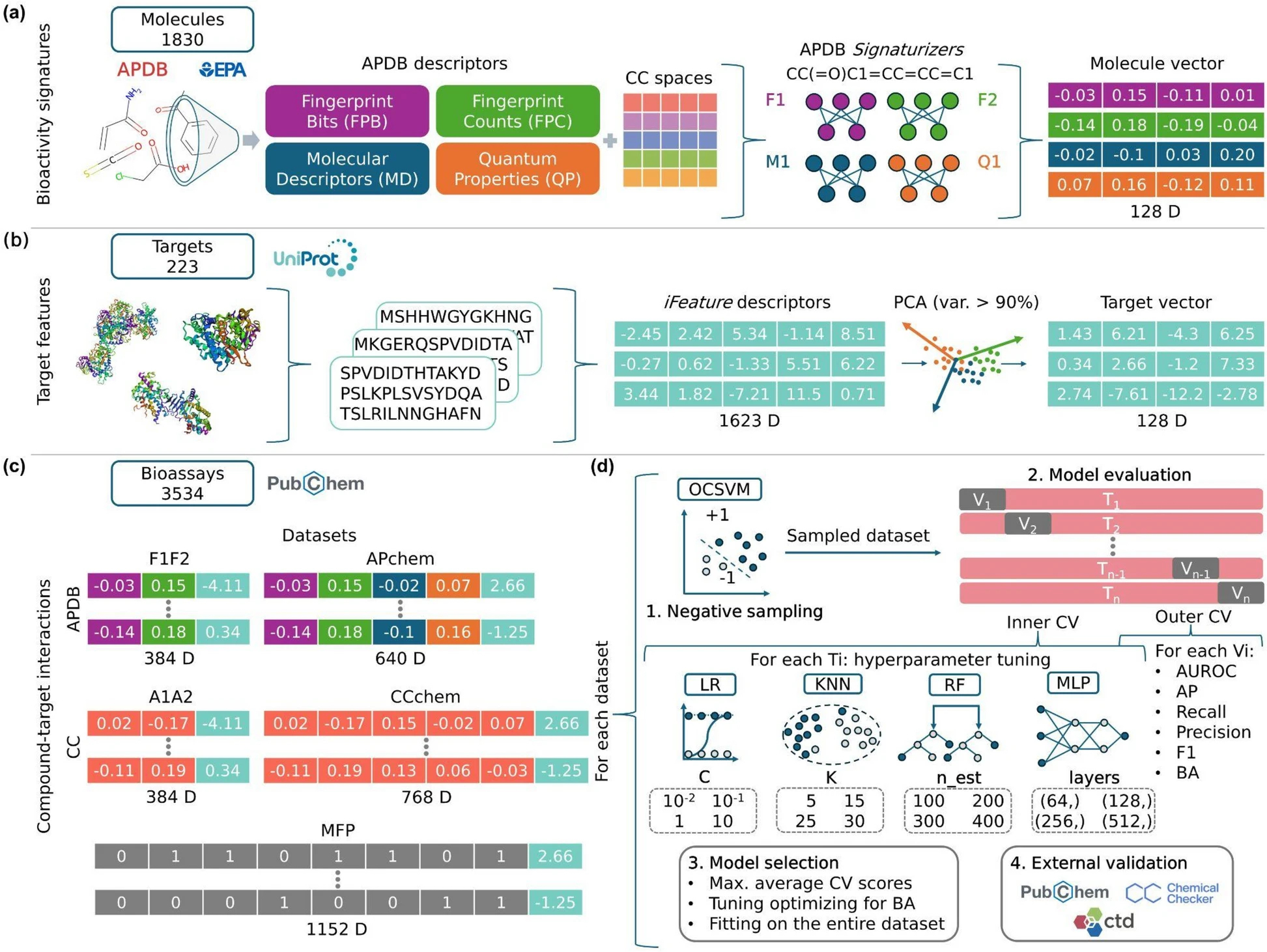
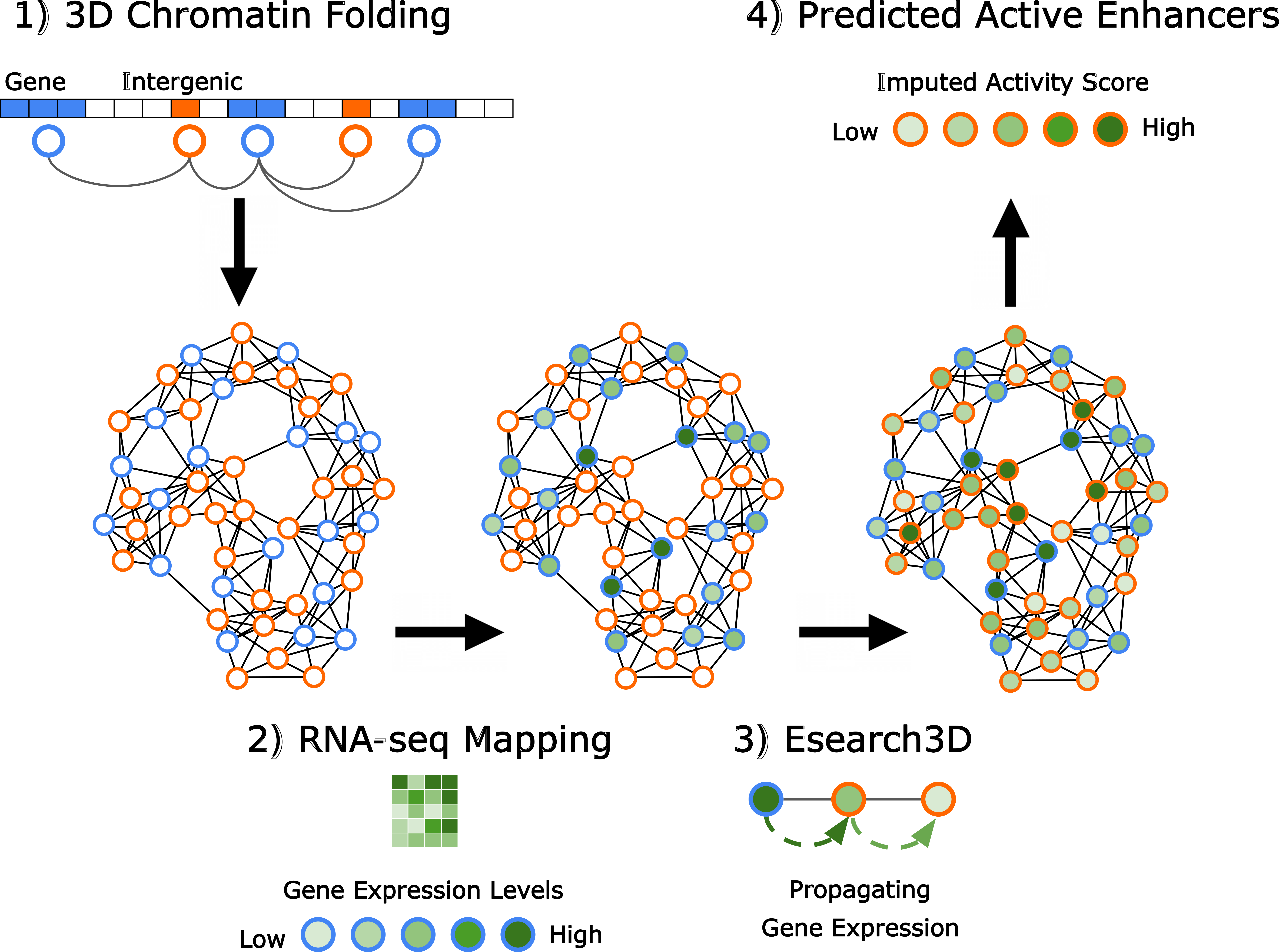
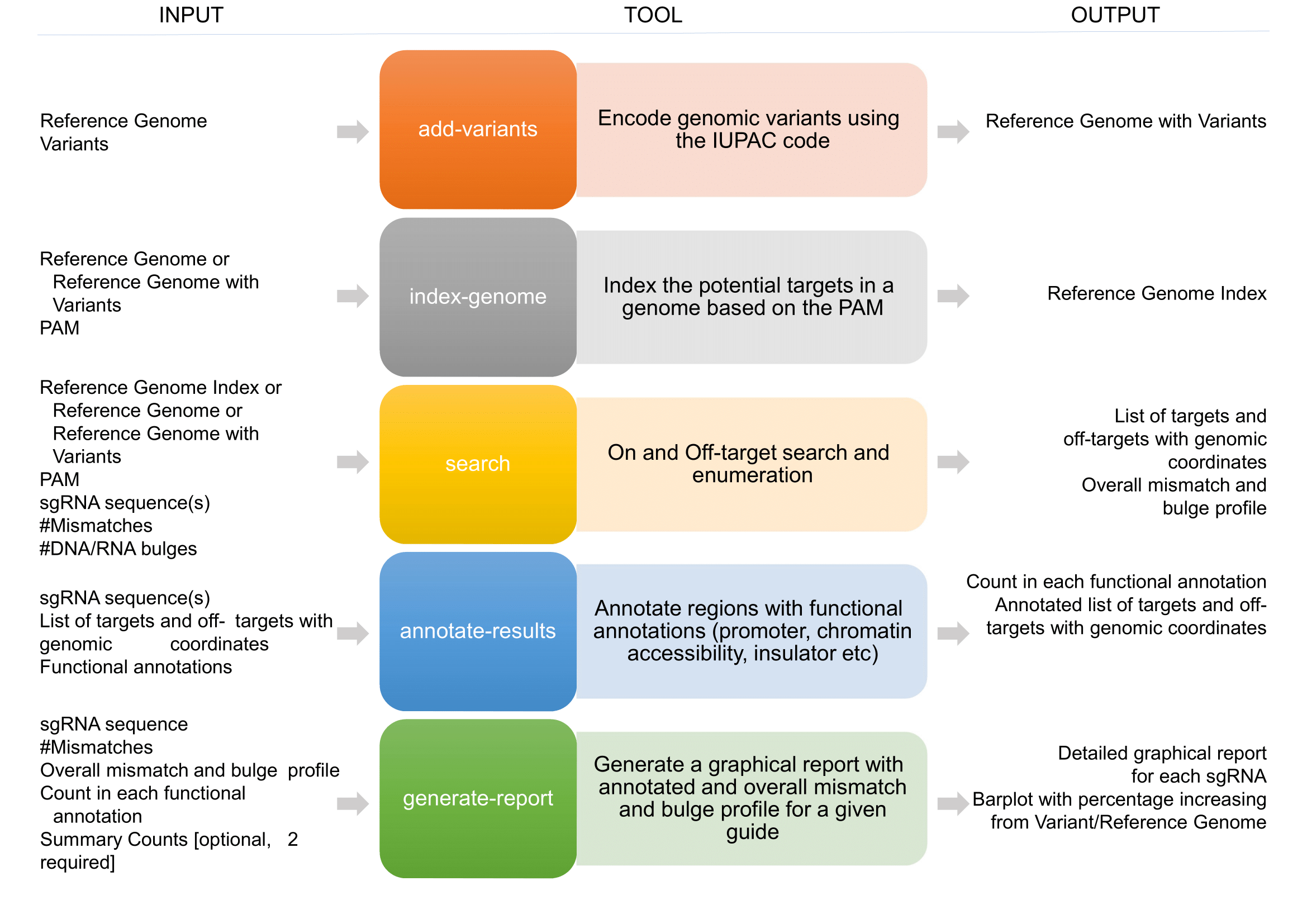
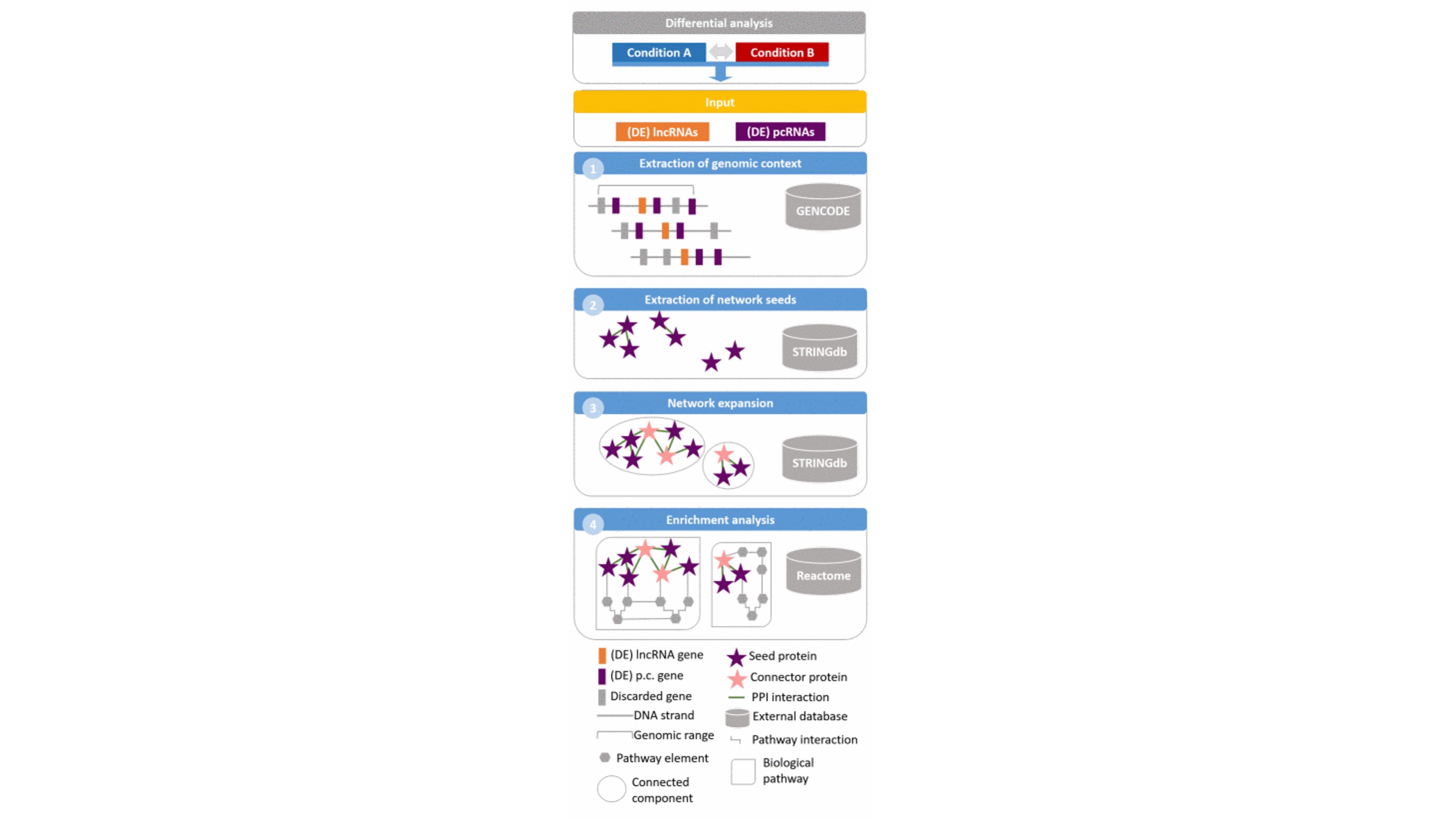
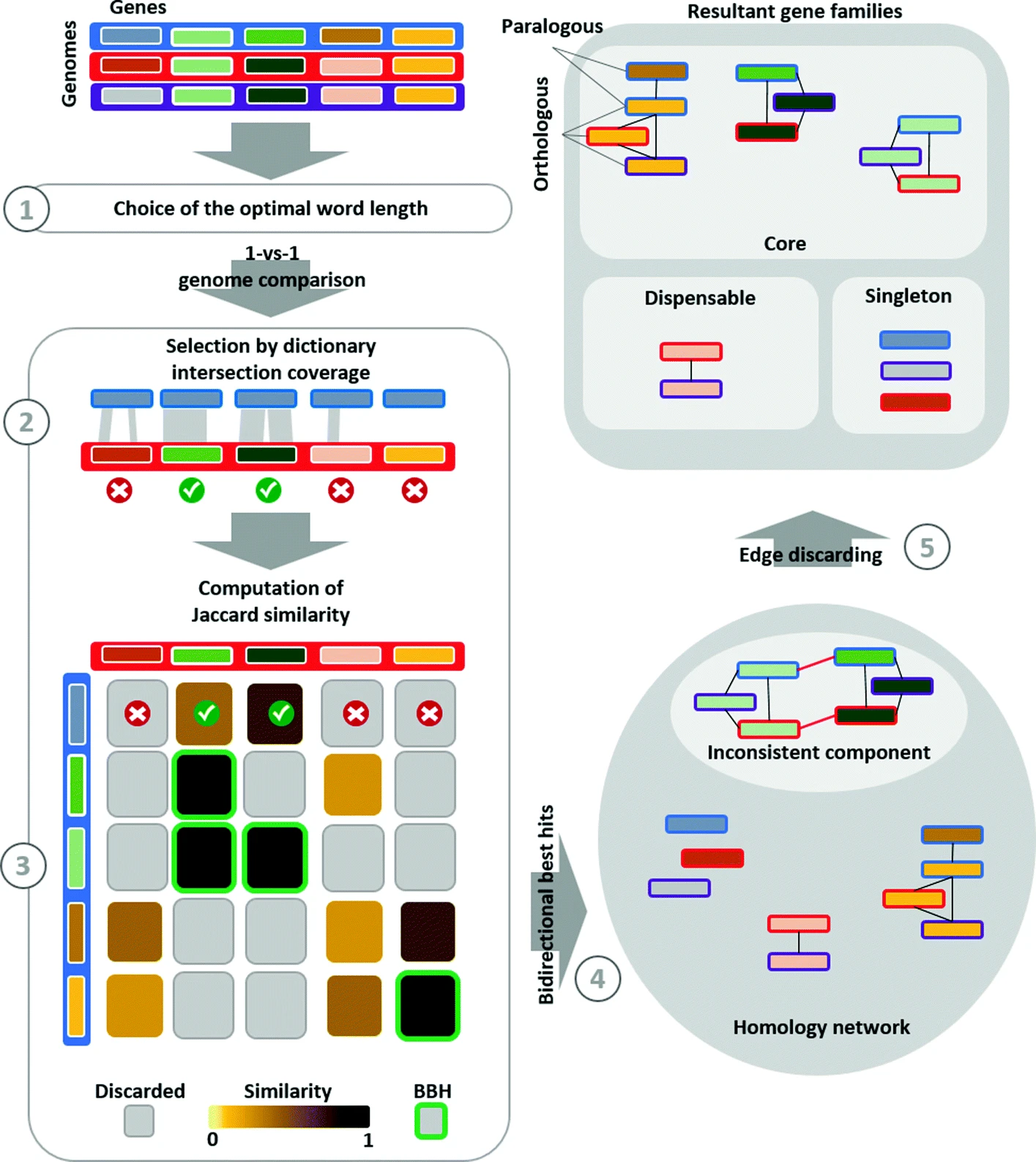
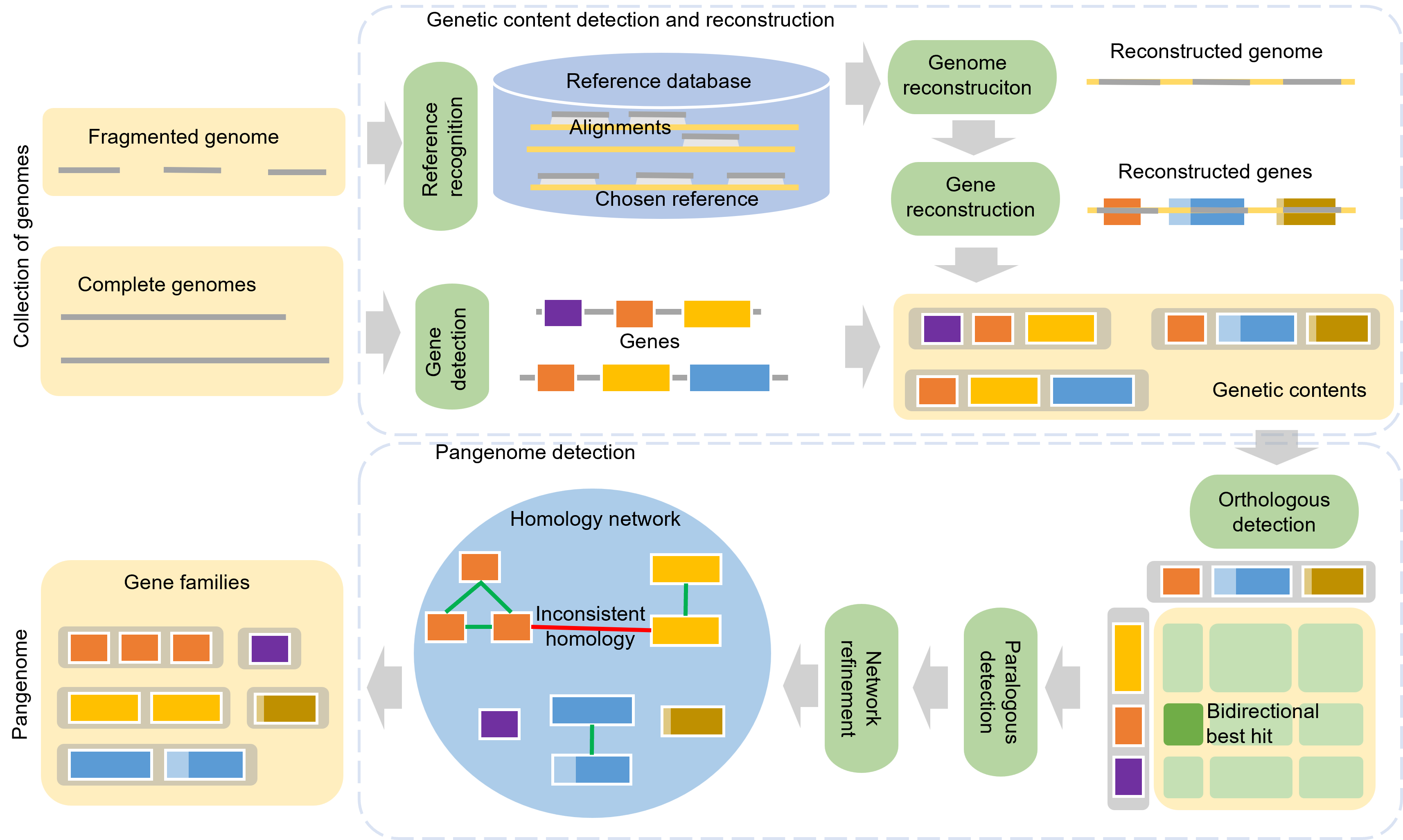
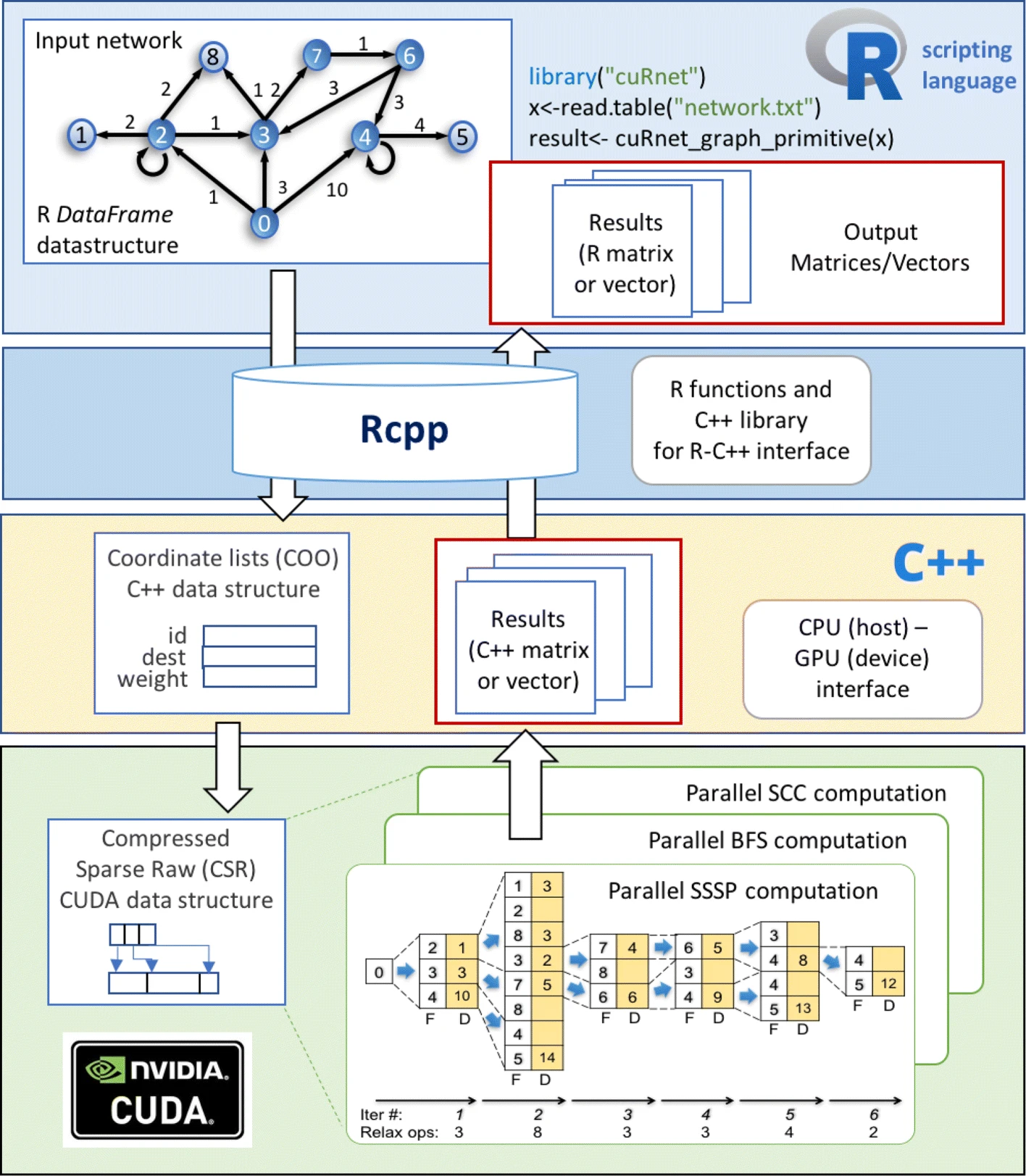
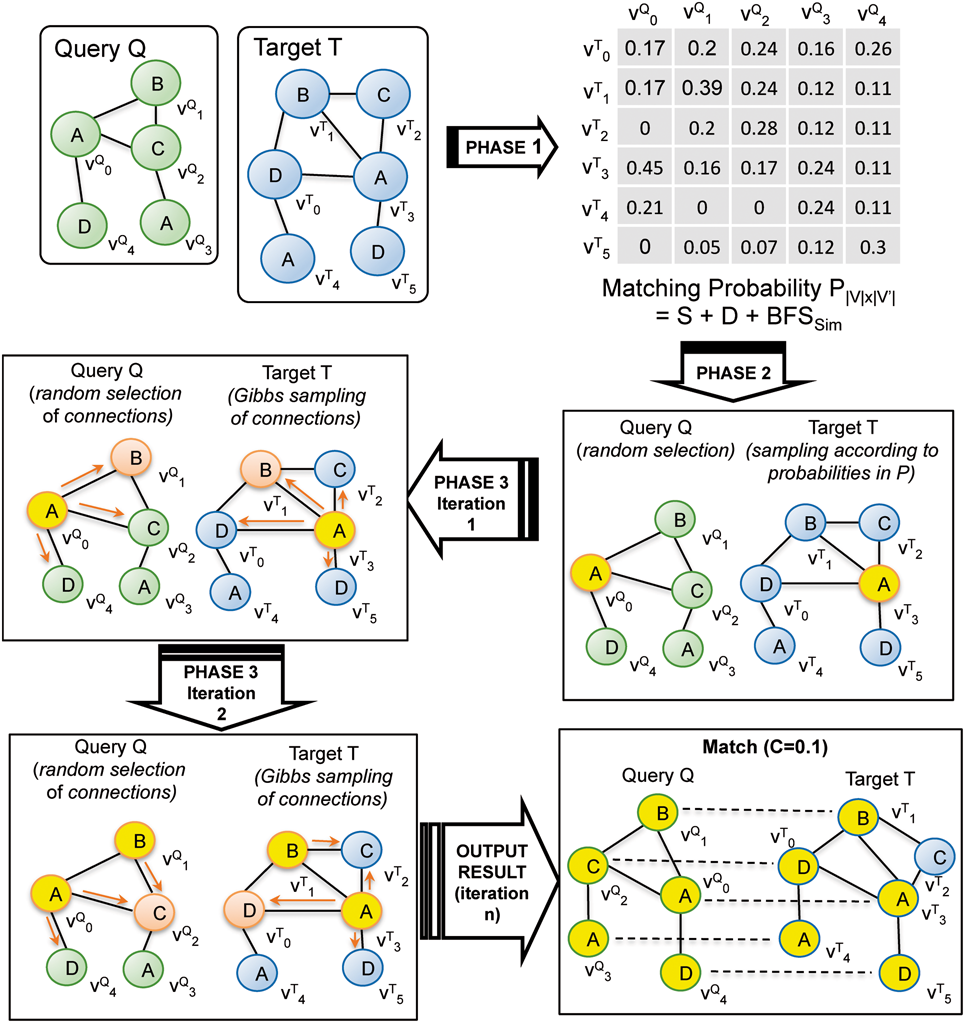
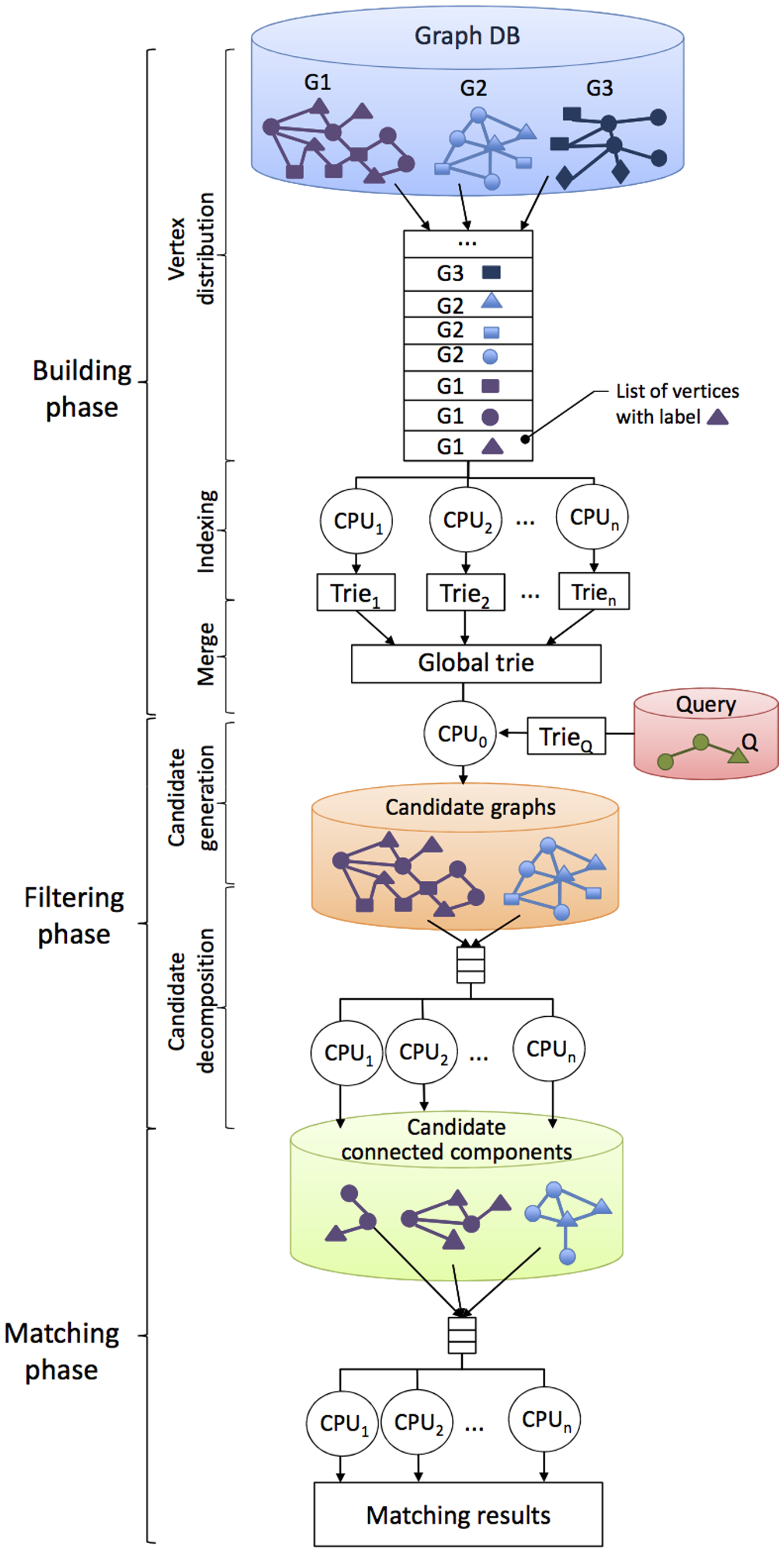
APBIO
APBIO is a tool developed to predict compound-target interactions, with a specific focus on air pollutants and their bioactivity. It computes bioactivity signatures for compounds starting from the SMILES representation (e.g., C1=CC=CC=C1) and FASTA sequence features for targets starting from the UniProtKB identifier (e.g., Q9UHW9). This allows unknown ligands to be prioritized for a given target (or vice versa), and subsequently tested through molecular docking simulations or validated via in vitro experiments for further studies on gene expression, chemical toxicity, and hazard evaluation.