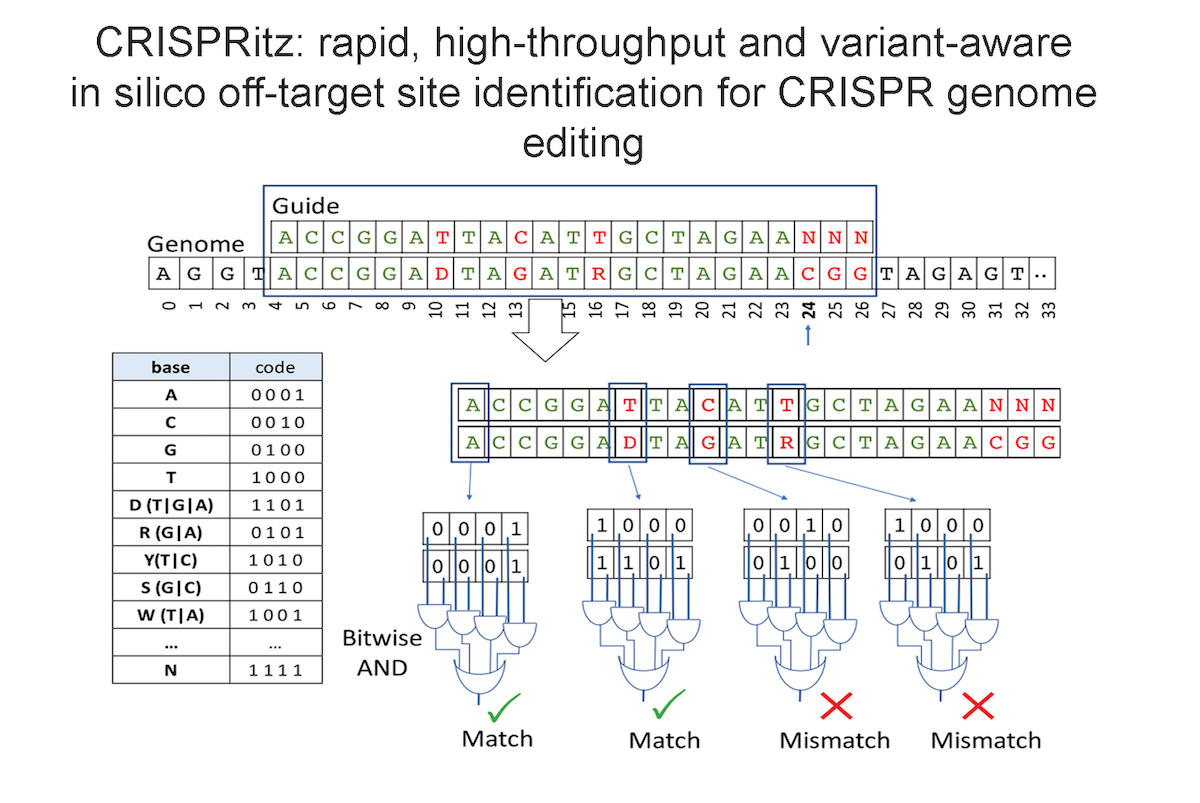
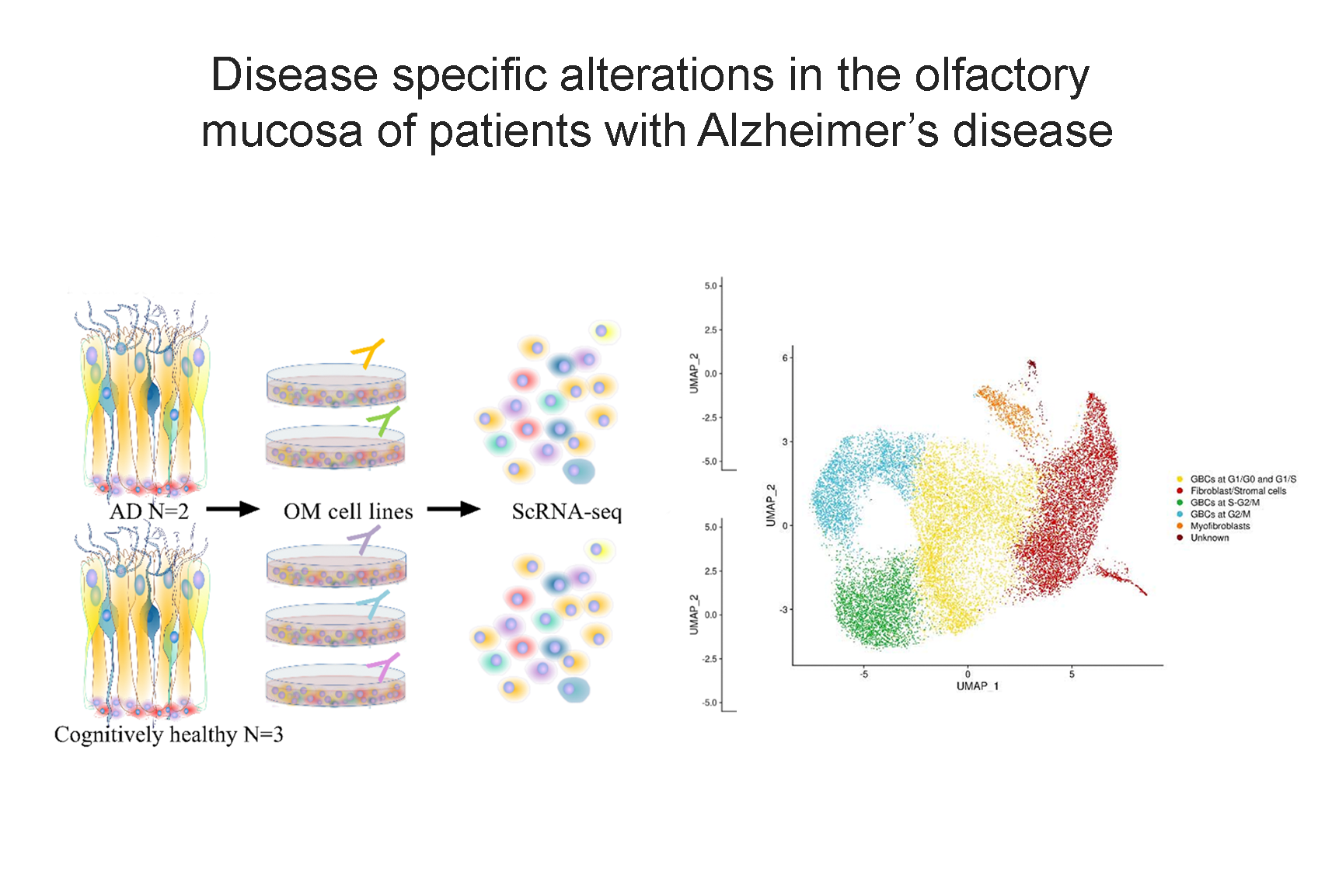
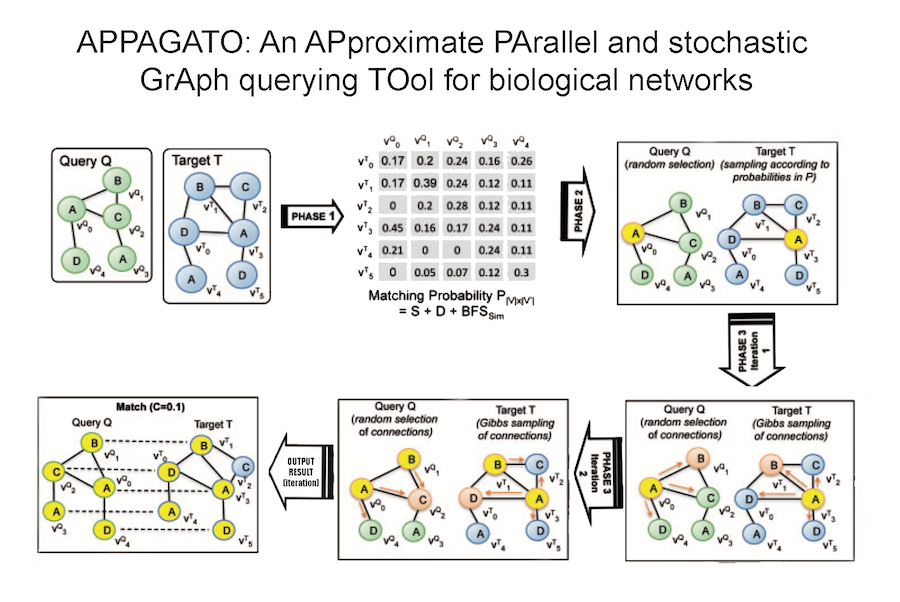
Our research aims to analyse biomedical data efficiently, in particular we develop new methods to mining biological networks, integrate heterogeneous data, analyse omics, reconstruct pangenomes, analyse genomes haplotype-aware and to classify patients. We use theory coming from machine learning, data science, mathematics and graph theory.